
Research Interests
Welcome to my realm of research—an enthralling journey where science meets innovation!
Within my research, I’ve dedicated myself to crafting theoretical and computational chemistry tools, unveiling the mysteries of physical chemistry in quantum materials and meta-materials. These explorations aren’t just about knowledge but about transforming the future. Imagine shaping cutting-edge technologies like emerging quantum advancements, revolutionizing energy solutions, and paving the way for sustainable progress.
My research unfolds in two compelling realms:
A. Method Developments: Think of this as building the foundation—creating powerful tools that decode the secrets of matter, empowering us to push boundaries and redefine possibilities.
B. Applications: Here’s where theory meets reality. I apply these tools to real-world challenges, channeling their potential to drive innovation across various domains—quantum technologies, sustainable energy, or beyond.
Thank you for joining me on this exhilarating expedition through the realms of science and discovery. Together, let’s delve deeper into the boundless potential of research!
A. Method Developments
I dive into the realm of computational methods right from the foundational bedrock—embracing ‘ab initio’ principles. Unlike theories that adapt to existing experimental data, I begin by solving the very fabric of quantum mechanics, unraveling the enigmatic Schrödinger Equations for nuclei and electrons, and plumbing the depths of statistical mechanics to compute materials’ properties.
These ‘first-principles’ theories offer a plethora of advantages. Picture this: they open doors to realms beyond experimental reach, shedding light on conditions that elude traditional measurement techniques. They become our guiding light, deciphering discrepancies between different samples or techniques, and even hold the key to architecting brand-new materials tailored for specific properties.
Yet, as with all brilliance, there are limitations. These theories demand computational prowess, and despite harnessing the mightiest supercomputers, their grasp is tethered by the constraints of time and scale. But fear not, for I wield a multi-scale approach—an intricate assembly of theories spanning hierarchies. This approach forms a symphony of knowledge, orchestrating a harmony that paints a vivid picture of these complex multi-scale processes. I engineer cutting-edge first-principle methods in the following areas:
1. Quantum vibronic effects
Picture a bustling city within a molecule or solid—where nuclei, like energetic dancers, pulse and sway around their equilibrium positions. Even at the frosty climes of absolute zero, quantum mechanics refuses to let them stand still, introducing what’s known as zero-point vibrations—an eternal dance dictated by quantum laws.
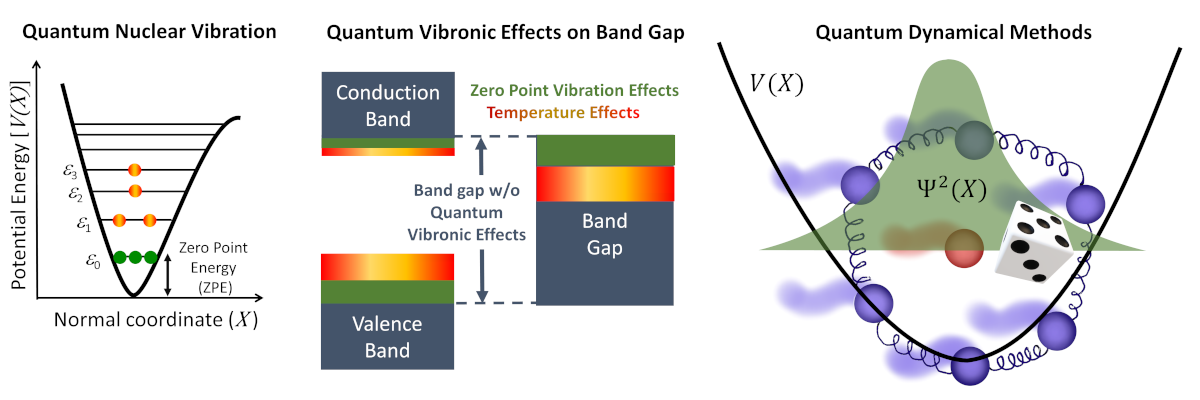
For ages, scientists overlooked this internal ballet, often modeling it with the simplicity of classical mechanics, while their focus remained fixated on solving the electronic Schrödinger equation. But here’s the twist: these quantum dance moves of the atomic nuclei hold profound power. They tug and reshape the electronic world, birthing the captivating tale of vibronic coupling—a phenomenon that reverberates across physics, chemistry, and the tech realms. Imagine this: from the fine-tuned symphonies of spectroscopy to the choreography of chemical reactivity and the vibrant canvases of photovoltaics and semiconductor technologies—vibronic coupling takes center stage, altering the very essence of these domains.
In my quest to reveal these hidden dynamics, I develop two distinctive quantum dynamical sampling techniques—(1) stochastic and (2) path-integral methods. These are not just tools; they’re portals allowing us to infuse quantum nuclear vibrations into the very fabric of electronic structure calculations. It’s the fusion of these worlds that propels us closer to decoding the quantum dance that shapes our materials and technologies.
2. Multi-scale ab-initio surface science modeling
Imagine the intricate interplay between a molecule and a surface—a duet that resonates across the vast realms of physics and chemistry. From catalytic wonders to the intricacies of gas technologies, from the artistry of thin films to the promise of nanotechnology—this partnership is the backbone of innovation. Unraveling the mysteries of these interactions, where surfaces whisper secrets to molecules, holds the golden key to unlocking groundbreaking technologies. Yet, it’s a labyrinthine journey, demanding simulations that retain microscopic resolution while spanning immense lengths and time scales. Enter my domain—a quest for ingenious multi-scale strategies.
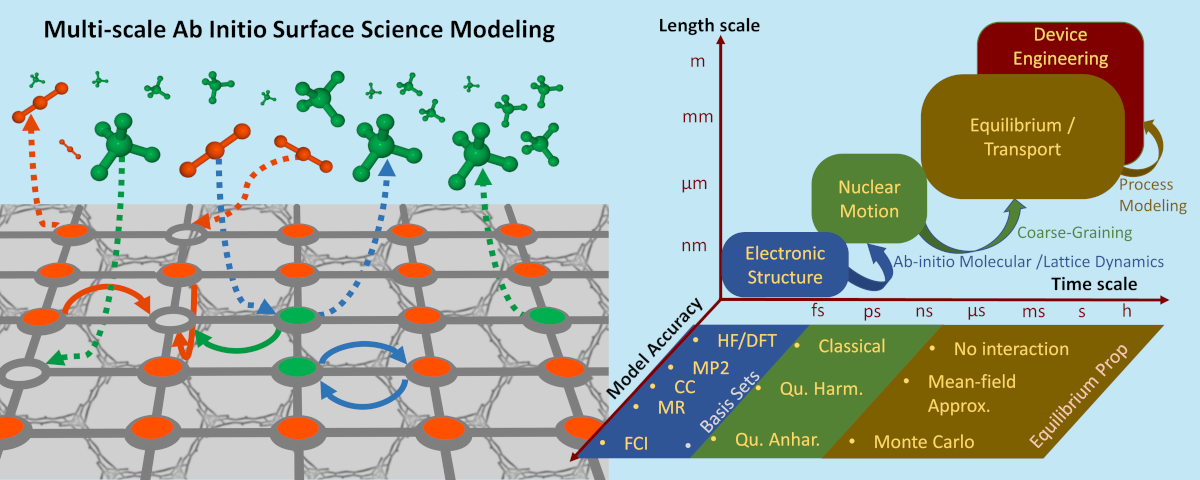
My arsenal? A fusion of quantum chemistry’s precision and statistical mechanics’ elegance. I navigate the electron’s universe with tools like density functional theory (DFT), Møller-Plesset (MP2) theory, and the advanced coupled cluster (CC) and multi-reference (MR) theories. But there’s more to this dance! To capture the nucleus’ heartbeat, I employ quantum models—harmonic and anharmonic—revealing the rhythm of molecular dynamics. Venturing beyond the atomic realm to the fascinating mesoscale, harnessing the power of precise Monte-Carlo simulations on coarse-grained models or cost-effective Mean-Field theories unveils the intricate unfolding of these processes. Behold the enigmatic artistry: within these multi-scale strategies, there’s no singular solution. No uniform concoction of hierarchical theoretical or computational tools can unveil the entirety of the properties we seek to understand.
3. AI-enhanced prediction of materials’ properties
Picture this: a bridge spanning the vast gulf between cutting-edge ab initio computational models and the sprawling landscapes of real-time processes. That’s precisely where the magic of artificial intelligence steps in. Here’s the thrill: sophisticated machine learning—armed with powerful algorithms—holds the key to shrinking the boundaries of time and length scales in computational modeling. But here’s the catch—quality data is the bedrock. Without it, the machine’s insight is but a mirage.
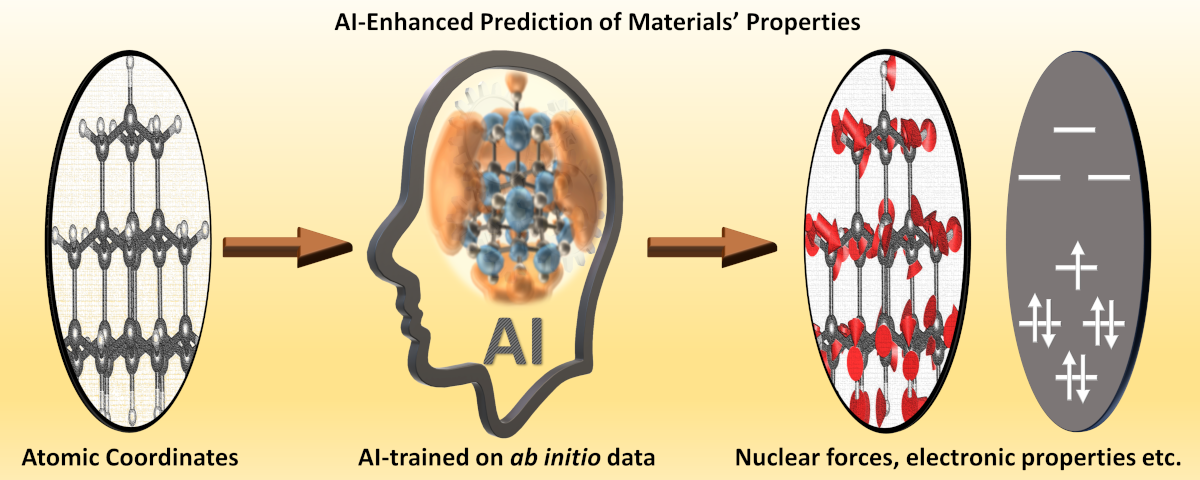
I thrive on the frontlines of this thrilling expedition: creating a goldmine of reliable ab initio data that fuels the learning engines of machine intelligence. And that’s just the beginning. I engineer the perfect language for these machines to comprehend—the right descriptors to unlock their true potential.
My canvas? Quantum materials—a realm where my machine learning models quickly produce ground and excited state forces on nuclei, electronic, and magnetic (spin) properties without the arduous journey through the Schrödinger equation. These models possess the prowess to unveil electronic properties, delving into spin dynamics that could reshape the landscape of spintronics, quantum information science, and the very fabric of quantum computing itself.
B. Applications
1. Materials Advancing Quantum Technologies
The landscape of quantum technologies brims with potential, from quantum sensing to communication and beyond. While current prototypes rely on superconducting spin-Qubits at cryogenic temperatures, the true frontier lies in unlocking their capabilities at room temperature—an innovation that fuels my passion.
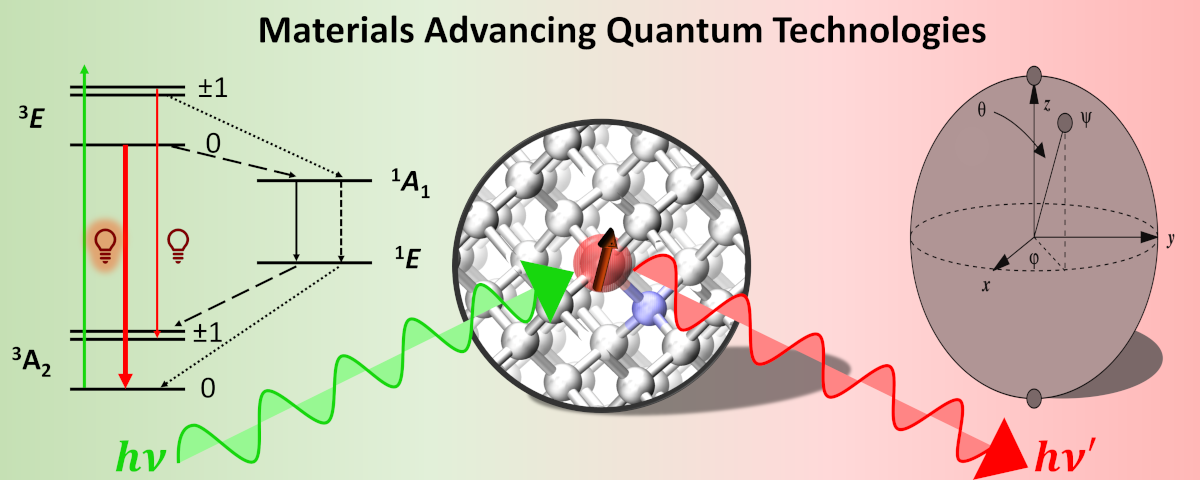
My focus? Semiconductor spin defects—these optically addressable spins offer a glimpse into room-temperature qubits. Yet, challenges abound. Extending spin coherence grapples with the elusive limits dictated by quantum vibronic effects.
I immerse myself in decoding the intricacies of optical spin detection, meticulously dissecting the nuances of spin-lattice and spin-spin interactions. From nitrogen-vacancy centers in diamond to divacancies in silicon carbide, these materials hold the promise of room-temperature quantum devices. But my journey doesn’t halt there—I venture into the domain of computer-assisted chemical modifications, paving the way for superior quantum materials.
2. Organic Electronic Materials
In the pursuit of a sustainable energy future, two critical fronts emerge: the clean energy generation, epitomized by solar power, and the design of eco-conscious, high-efficiency electronics. Step onto the scene: organic molecules, polymers, and carbon nanomaterials—a trio at the forefront of this pursuit. Not only environmentally friendly, these materials boast electronic adaptability, fine-tuning their capabilities through meticulous packing or subtle chemical tweaks.
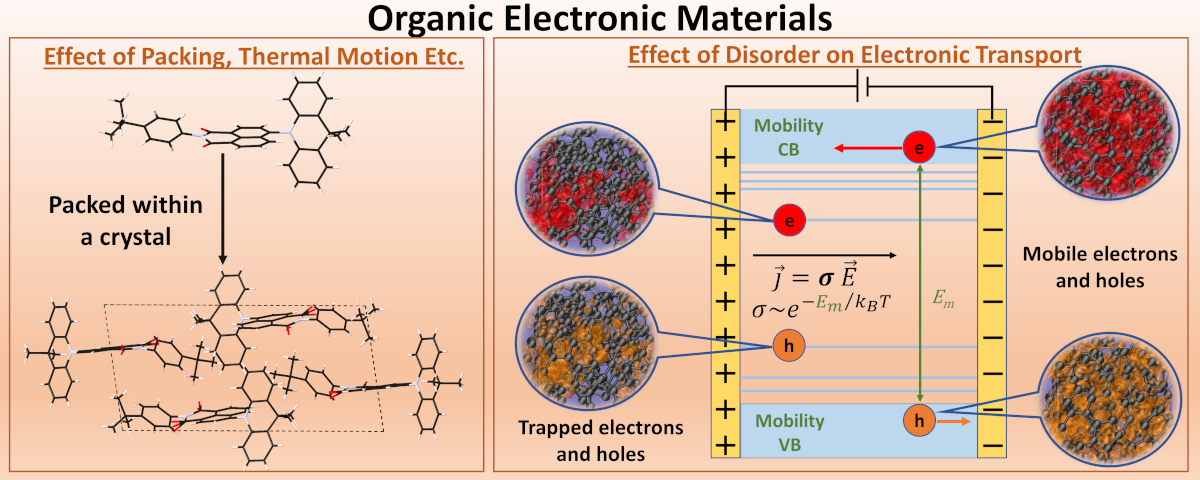
However, the path to efficiency is a winding one. Hindered by short exciton diffusion lengths and subpar carrier mobilities due to inherent static and dynamic disorders, achieving peak performance remains elusive. Among the myriad possibilities, the most efficient packing arrangement demands an extensive exploration.
Armed with first-principle methods, I embark on unraveling these mysteries, seeking solutions to these challenges that stand between us and a greener, more efficient tomorrow.
3. Gas Separation & Storage using Porous Materials
Gas separation and storage applications are pivotal in addressing sustainability challenges—separating CO2 from flue gas, storing energy-rich molecules like CH4 or H2, producing drinking water via humidity capture, and even in gas mask development. Porous crystalline solids, namely metal-organic frameworks (MOFs) and covalent organic frameworks (COFs), possess high porosity, extensive surface areas, and adaptable cross-linking capabilities. These materials can be precisely tailored to meet specific gas separation and storage needs.
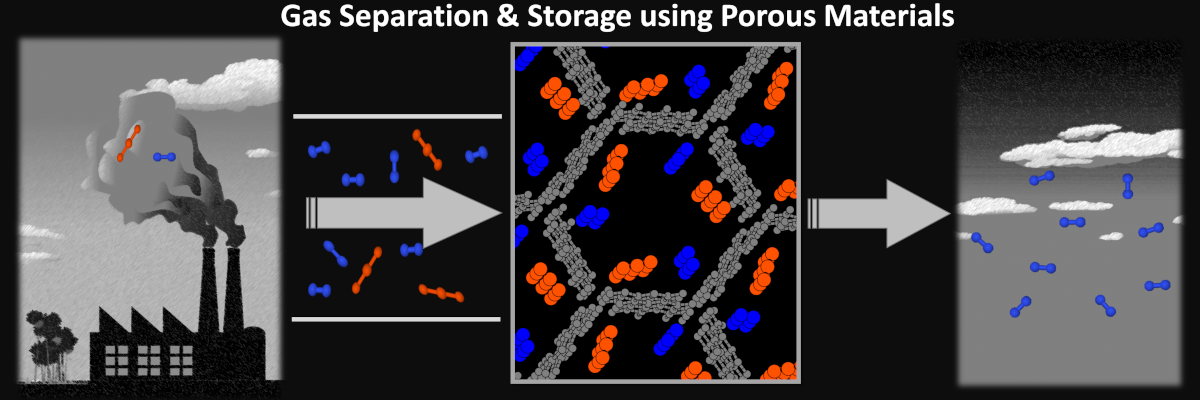
Assessing the working conditions necessary for a particular MOF or COF in addressing a gas separation challenge becomes a crucial determinant of its synthesis viability. Therefore, employing multi-scale simulation methodologies becomes essential. These simulations delve into the physicochemical processes, exploring co-adsorption equilibria and gas diffusion within the pores. Ultimately, my goal is to weave a comprehensive thread of knowledge, bridging the intricacies of chemical bonds to the practical applications in chemical plants.